
Коэнзим Q10 (убихинон) в настоящее время рекомендован в качестве дополнения к традиционной терапии сердечно–сосудистых заболеваний [1,7,9]. Хотя коэнзим Q10 является липофильным соединением, растворимость его крайне ограничена и препараты часто характеризуются низкой биодоступностью [8]. На степень всасывания вещества наибольшее влияние оказывают физико–химические свойства его субстанции в составе препарата, следовательно, коэнзим Q10 в порошке, суспензии, масляном растворе, солюбилизированной форме и т.д., скорее всего, будет демонстрировать разную биодоступность. Однако исследований фармакокинетических характеристик субстанции и препаратов коэнзима Q10 чрезвычайно мало.
Целью данной работы явилось изучение фармакокинетики порошка и раствора солюбилизированной формы коэнзима Q10 после однократного перорального введения крысам.
Методы исследования
Экспериментальные животные и хирургические процедуры
Исследование выполнено на самцах крыс линии Wistar массой 250–300 г. Животным в стерильных условиях под наркозом (пентобарбитал, 50 мг/кг внутрибрюшинно) имплантировали катетер в бедренную вену для последующего отбора образцов крови. Через сутки после операции бодрствующим животным натощак через зонд вводили в желудок коэнзим Q10 в дозе 10 мг/кг: крысам первой группы (n=7) – раствор солюбилизированного коэнзима Q10 («Кудесан», ЗАО «АКВИОН», Москва, Россия), крысам второй группы (n=7) – порошок коэнзима Q10 в виде взвеси в 0,2% растворе метилцеллюлозы.
Отбор проб крови (по 0,3 мл) проводили до введения препарата и спустя 2, 3, 4, 5, 7, 9, 24, 36 и 48 ч после введения. Образцы крови центрифугировали, плазму крови отбирали, замораживали и хранили при –20°С до проведения количественного анализа коэнзимов Q. На протяжении двух суток после введения коэнзима Q10 животных не кормили, не ограничивая доступ к воде.
Определение коэнзима Q10
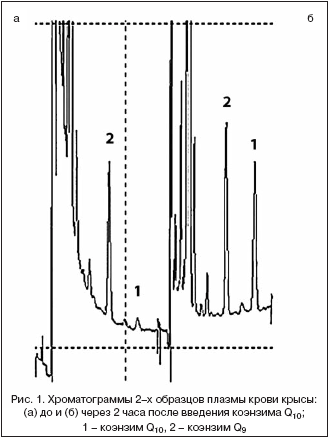
Количественный анализ коэнзима Q10 в плазме крови проводили по методике [5] с некоторыми модификациями. К 100 мкл плазмы крови добавляли 220 мкл этанола и 550 мкл n–гексана, тщательно встряхивали в течение 10 мин, центрифугировали при 3000 об/мин в течение 3 мин и верхний слой n–гексана отбирали в объеме 500 мкл. К остатку добавляли еще 550 мкл n–гексана и повторяли процедуру экстракции и отбора экстракта. Объединенный экстракт упаривали досуха, растворяли в 100 мкл этанола и восстанавливали окисленную форму коэнзимов Q9 и Q10 добавлением 10 мкл 5% раствора натрия тетрагидробората в этаноле. 10 мкл восстановленного экстракта анализировали с помощью высокоэффективной жидкостной хроматографии с электрохимическим детектированием на оборудовании фирмы «Environmental Sciences Associate, Inc.» (США) – насос модели 580 и электрохимический детектор «Coulochem II». Условия проведения хроматографического анализа обеспечивали разделение коэнзимов Q9 и Q10, необходимое ввиду того, что доминирующей формой коэнзима Q у крыс является коэнзим Q9 (рис. 1). Разделение проводили в изократическом режиме на колонке 150х4,6 мм с сорбентом С18 (5 мкм) при скорости потока элюэнта 1,3 мл/мин. Подвижная фаза содержала 0,3% NaCl в смеси этанол: метанол: 7% HClO4 (970:20:10). Электрохимическое детектирование осуществляли с помощью аналитической ячейки (model 5011) при напряжении на первой паре электродов –50 мВ и +350 мВ – на второй. Время удерживания составило для коэнзимов Q9 и Q10 – 6,5 и 8,5 мин соответственно. Регистрацию и обработку хроматографических данных проводили с помощью компьютерной программы фирмы «Environmental Sciences Associate, Inc.» (США).
Определение фармакокинетических характеристик коэнзима Q10
Для порошка и солюбилизированной формы коэнзима Q10 были определены и рассчитаны следующие параметры фармакокинетики: Tmax – время достижения максимальной концентрации, Cmax – величина максимальной концентрации, AUCo–t – площадь под фармакокинетической кривой “концентрация–время”, Cmax/AUCo–t – относительная скорость всасывания. Для солюбилизированной формы рассчитывали: f – относительную биодоступность исследуемой лекарственной формы по отношению к сравниваемой, определяемую отношением AUCo–t,T/AUCo–t,R, f’ – относительную степень всасывания, определяемую отношением Cmax,T/Cmax,R.
Значения параметров фармакокинетики были рассчитаны модельно–независимыми методами. Величину Cmax и время ее достижения определяли из фактических значений концентраций. Площадь под фармакокинетической кривой (AUCo–t) рассчитывали методом трапеций.
Расчет биодоступности коэнзима Q10 производился методом сравнения площадей под кривыми зависимости концентрации коэнзима Q10 в плазме крови от времени, построенными для двух групп крыс.
Обработка результатов. Результаты представлены как среднее значение ± стандартное отклонение (M±m). Межгрупповые различия оценивали, используя t–test. Различия считали значимыми при вероятности ошибки менее 5% (p<0,05).
Результаты исследования
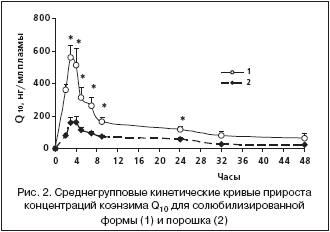
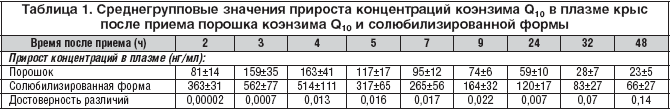
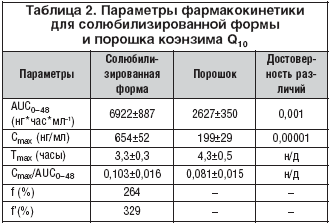
Полученные величины времени достижения максимальной концентрации коэнзима Q10 в плазме (3–4 ч) свидетельствуют о медленной абсорбции коэнзима Q10 в желудочно–кишечном тракте вследствие его гидрофобности и большого молекулярного веса. Время достижения максимальной концентрации и относительная скорость всасывания порошка коэнзима Q10 и солюбилизированной формы достоверно не различались. Другие фармакокинетические параметры коэнзима Q10 в солюбилизированной форме существенно отличались от таковых в порошке (рис. 2). Средние значения концентраций коэнзима Q10 в плазме в течение суток после приема солюбилизированной формы были достоверно выше, чем после приема порошка (табл. 1). Максимальная концентрация в плазме крови после приема солюбилизированной формы была примерно в 3,3 раза выше, а площадь под кривой концентрация–время – в 2,6 раза больше по сравнению с порошком (табл. 2). Соответственно, биодоступность коэнзима Q10 в составе солюбилизированной формы относительно порошка составляла 264%, а относительная степень всасывания 329% (табл. 2).
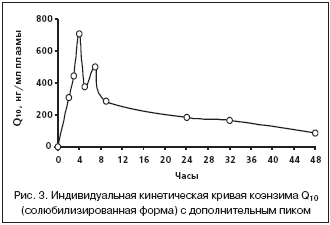
Кинетические кривые коэнзима Q10 у нескольких животных имели дополнительные пики концентраций, наблюдавшиеся в интервале от 7 до 24 часов после введения вещества (рис. 3). Известно, что жирорастворимый коэнзим Q10 после всасывания в кишечнике захватывается из циркуляции тканями печени [3,13]. По–видимому, наличие дополнительных пиков концентраций коэнзима Q10, наблюдавшееся также в исследованиях на людях [10] и на морских свинках [11], является отражением феномена энтерогепатической циркуляции. Эпизоды повторного всасывания коэнзима Q10 обусловили ломаный характер кинетических кривых после максимума концентраций и исключили возможность корректного расчета константы элиминации.
Таким образом, коэнзим Q10 в солюбилизированной форме обладает несомненными преимуществами: большей степенью всасывания, более высокими концентрациями в плазме крови и в результате – большей биодоступностью. Это согласуется с имеющимися в литературе данными, показывающими, что при длительном приеме солюбилизированных форм плазменные концентрации коэнзима Q10 в 2–2,5 раза выше [4], а относительная биодоступность в 3–6 раз больше, чем при использовании порошка [6,12]. Фармакокинетические преимущества солюбилизированной формы обусловили ее высокую эффективность в качестве кардиопротектора: хроническое пероральное введение приводило к повышению не только плазменных уровней коэнзима Q10 (в 2,5 раза), но и содержания его в миокарде крыс, что увеличивало выживаемость клеток миокарда в условиях ишемии и в итоге ограничивало размер постинфарктной зоны некроза [2].
Литература
1. Аронов Д.М. // Русский медицинский журнал. 2004. Т.12. № 15. С. 905–909.
2. Каленикова Е.И., Городецкая Е.А., Колокольчикова Е. и др. // Биохимия. 2007. Т. 72. № 3. С. 407–415.
3. Bhagavan H.N., Chopra R.K. // Free Radical Res. 2006. V. 40. P. 445–453.
4. Chopra R.K., Godman R., Sinatra S.T., and Bhagavan H.N. // Int. J. Vitam. Nutr. Res. 1998. V. 68. № 2. P.109–113.
5. Lass A., and Sohal R. // Free Radic. Biol. & Med. 1998. V. 27. P. 220–226.
6. Miles M., Horn P., Miles L. et al. // Nutr. Res. 2002. V. 22. P. 919–929.
7. Sarter B. // J. Cardiovasc. Nurs. 2002. V. 16. P. 9–20.
8. Ullman U., Metzner J., Schulz C. et al. // Journal of Medical Food. 2005. V.8. № 3. P. 397–399.
9. Weant K.A., and Smith K.M. // Ann. Pharmacother. 2005. V. 39. P. 1522–1526.
10. Weiss M., Mortensen S.A., Rassing M.R. et al. // Mol. Aspects Med. 1994. V. 15. P. s273–s280.
11. Yuzuriha T., Takada M., Katayama K. // Biochim. Biophys. Acta. 1983. V. 759. P. 286–291.
12. Zaghloul A., Gurley B., Khan M. et al. // Drug. Dev. Ind. Pharm. 2002. V. 28. № 10. P. 1195–1200.
13. Zhang Y., Aberg F., Appelkvist E–L. et al. // J. Nutr. 1995. V. 125. P. 446–453.
Источник: rmj.ru